Al decennialang proberen evolutiebiologen de levensboom te reconstrueren. Hoe gaan ze precies te werk?

Cladistiek
De Duitse entomoloog Willi Hennig introduceerde cladistiek om evolutionaire bomen te bouwen. Cladistiek gebruikt de uiterlijke kenmerken van diverse groepen organismen. Welke kenmerken worden gedeeld door verschillende organismen? En welke kenmerken zijn uniek voor een bepaalde groep? Op basis van de verdeling van deze kenmerken kun je vervolgens een evolutionaire boom tekenen. Met deze methode kwam ook een karrenvracht aan nieuwe tongbrekende termen, zoals symplesiomorfie en autapomorfie. Vele van deze termen worden nog steeds gebruikt, daarom neem ik even de tijd voor een snelcursus cladistiek. Als voorbeeld neem ik de gewervelden, dieren met een ruggenmerg. Ik kies een vertegenwoordiger voor elke diergroep en ik maak een tabel waarin ik de aan- en afwezigheid van bepaalde kenmerken aangeef. Bijvoorbeeld, bijna alle gewervelden hebben een benig kalkskelet. Haaien hebben echter een skelet dat is opgebouwd uit kraakbeen. Daarom geef ik alle gewervelden de score 1 en haaien de score 0 voor dit kenmerk. Met deze tabel of matrix kan ik vervolgens dieren gaan groeperen op basis van de kenmerken die ze delen. Het eindresultaat is onderstaand cladogram. De twijgjes van deze boom zijn de nog levende organismen, terwijl de knooppunten in de boom uitgestorven voorouders vertegenwoordigen.
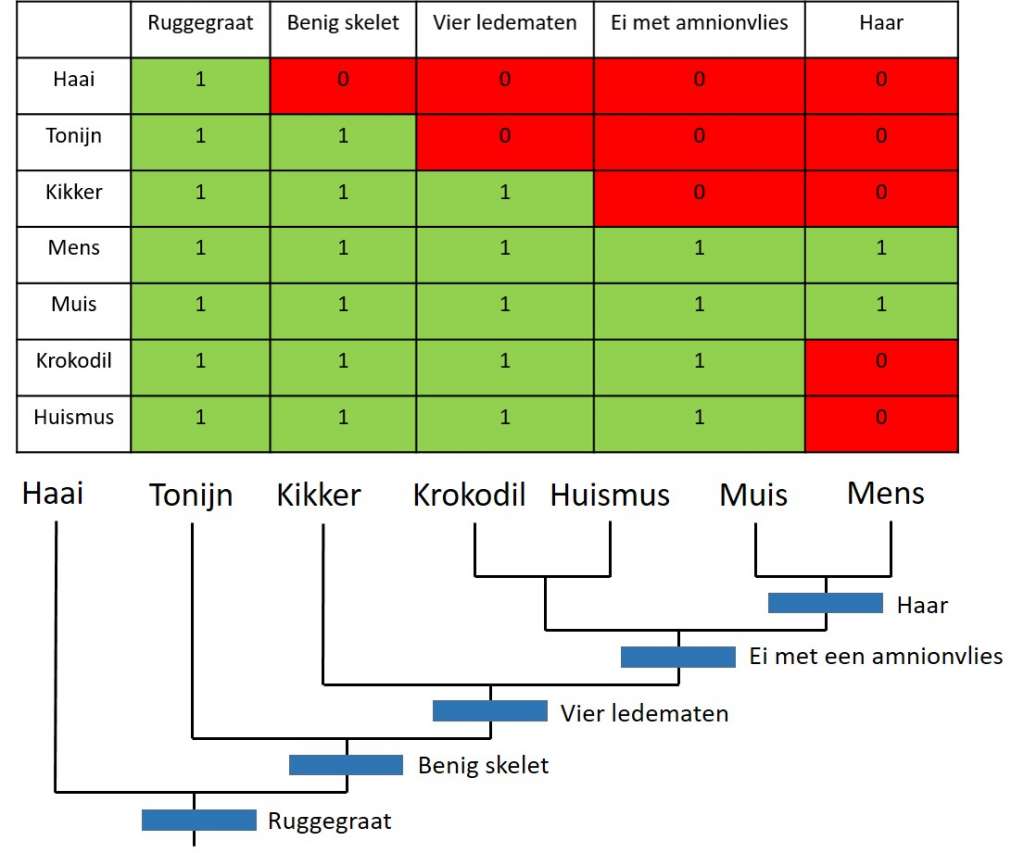
Apomorfie en homoplasie
Een nieuw geëvolueerd kenmerk dat verschilt van de voorouder wordt een apomorfie genoemd. Wanneer verschillende leden van een groep ditzelfde kenmerk delen, dan spreken we van een synapomorfie (syn- staat voor samen). De aanwezigheid van haar bij de muis en de mens is een voorbeeld van een synapomorfie. Hoe meer synapomorfieën een bepaalde groep deelt, hoe betrouwbaarder het cladogram. Voor zoogdieren is er een hele waslijst aan apomorfieën bekend: denk aan het voeden van jongen met moedermelk of warmbloedigheid. Bij de oplettende lezer zou nu een alarmbel moeten rinkelen. Warmbloedigheid een uniek kenmerk voor zoogdieren? Vogels zijn toch ook warmbloedig? Moeten vogels en zoogdieren dan niet in dezelfde groep ondergebracht worden? Neen, want er zijn talloze andere kenmerken die aantonen dat vogels en zoogdieren tot aparte evolutionaire groepen behoren. Warmbloedigheid is een voorbeeld van een zogenaamde homoplasie, een kenmerk dat gedeeld wordt door organismen van totaal verschillende groepen. Vogels en zoogdieren zijn onafhankelijk van elkaar warmbloedig geworden. Andere voorbeelden van een homoplasie zijn de vleugels van vogels en vleermuizen of de gestroomlijnde vorm van vissen en dolfijnen. Dit voorbeeld geeft aan dat cladogrammen gebaseerd moeten zijn op zo veel mogelijk kenmerken. Als een onderzoeker te weinig kenmerken gebruikt en daar zitten ook nog eens een paar homoplasieën tussen, dan zal hij de verkeerde conclusies trekken. In mijn voorbeeld gebruikte ik slechts een handvol kenmerken, maar in wetenschappelijke studies worden honderden kenmerken met elkaar vergeleken. Bijvoorbeeld, in een recente studie naar de evolutie van uitgestorven loopvogels, analyseerde Trevor Worthy (Flinders Universiteit, Adelaide, Australië) met zijn collega’s bijna 300 verschillende kenmerken.

De mogelijkheid om het genetisch materiaal van een individu af te lezen zorgde voor een revolutie in het onderzoek naar evolutionaire stambomen. Plots had men DNA-sequenties, opgebouwd uit de letters A, T, G en C, ter beschikking. Het vergelijken van deze sequenties vroeg wel om de ontwikkeling van nieuwe methoden en software. Ik zal enkele van deze methoden uitleggen aan de hand van DNA-sequenties van mens, chimpansee en gorilla.
Spaarzaamheid
Een eerste methode is gebaseerd op parsimonie (of spaarzaamheid), de aanname dat als er meerdere verklaringen zijn, de eenvoudigste de voorkeur krijgt. Onderzoekers gaan op zoek naar maximale parsimonie (meestal afgekort als MP). In het geval van evolutionaire bomen, betekent dit het minste veranderingen in DNA-sequenties. Met andere woorden, zo weinig mogelijk mutaties. Als je de DNA-sequenties van de drie mensapen vergelijkt, dan heeft de simpelste evolutionaire boom slechts twee mutaties nodig. Een mutatie van T naar C (in het blauw) en een mutatie van C naar A (in het rood). De belangrijkste kritiek op deze methode is de aanname dat evolutie de meest spaarzame weg kiest. Het is niet altijd zeker dat de kortste weg (met het minste mutaties) ook de weg is die evolutie gevolgd heeft.
Genetische afstand
Een andere manier om een evolutionaire boom af te leiden uit DNA-gegevens is met behulp van genetische afstanden. Simpel gezegd, je berekent het verschil in DNA-sequentie tussen alle soorten. In mijn mensapenvoorbeeld is de genetische afstand tussen mens en chimpansee ongeveer acht procent (één letter verschil in een sequentie van twaalf letters). Het verschil tussen mens en gorilla is zestien procent (twee letters verschil). De mens staat dus genetisch dichter bij de chimpansee dan bij de gorilla. In deze berekening ga ik ervan uit dat elke mutatie even waarschijnlijk is, maar dat blijkt niet altijd het geval te zijn. Sommige mutaties komen vaker voor dan andere. Dit leidde tot de ontwikkeling van diverse mutatiemodellen.
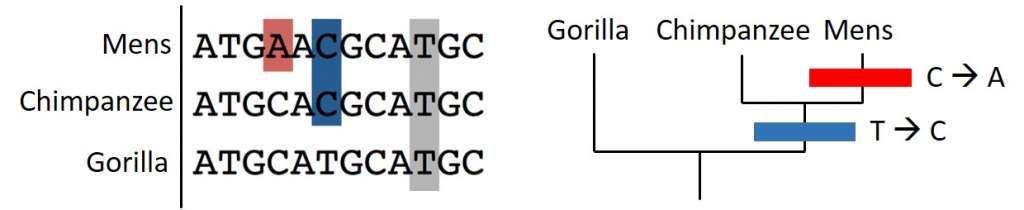
Mutatiemodellen
Het simpelste mutatiemodel – dat ik ook gebruikte in het voorbeeld – werd uitgewerkt door Thomas Jukes en Charles Cantor in 1969 (in de literatuur staat het bekend als JC69). In dit model komt elke mutatie met dezelfde kans voor. In 1980 introduceerde Motoo Kimura een extra wiskundige parameter, namelijk de chemische structuur van de DNA-letters. A en G behoren tot dezelfde chemische groep (de purines), terwijl C en T tot een andere groep behoren (de pyrimidines). Mutaties tussen letters van dezelfde chemische groep zijn waarschijnlijker. Bijvoorbeeld, de mutatie van A naar G (beide purines) komt vaker voor dan een mutatie van A naar T (van purine naar pyrimidines). Ondertussen zijn er diverse mutatiemodellen geïntroduceerd. Ik zal je de details besparen, maar nieuwsgierige lezers met een wiskundeknobbel kunnen zoeken naar de modellen van Joseph Felsenstein (F81), Hasegawa, Kishino en Yano (HKY85), of Tamura en Nei (TN93).
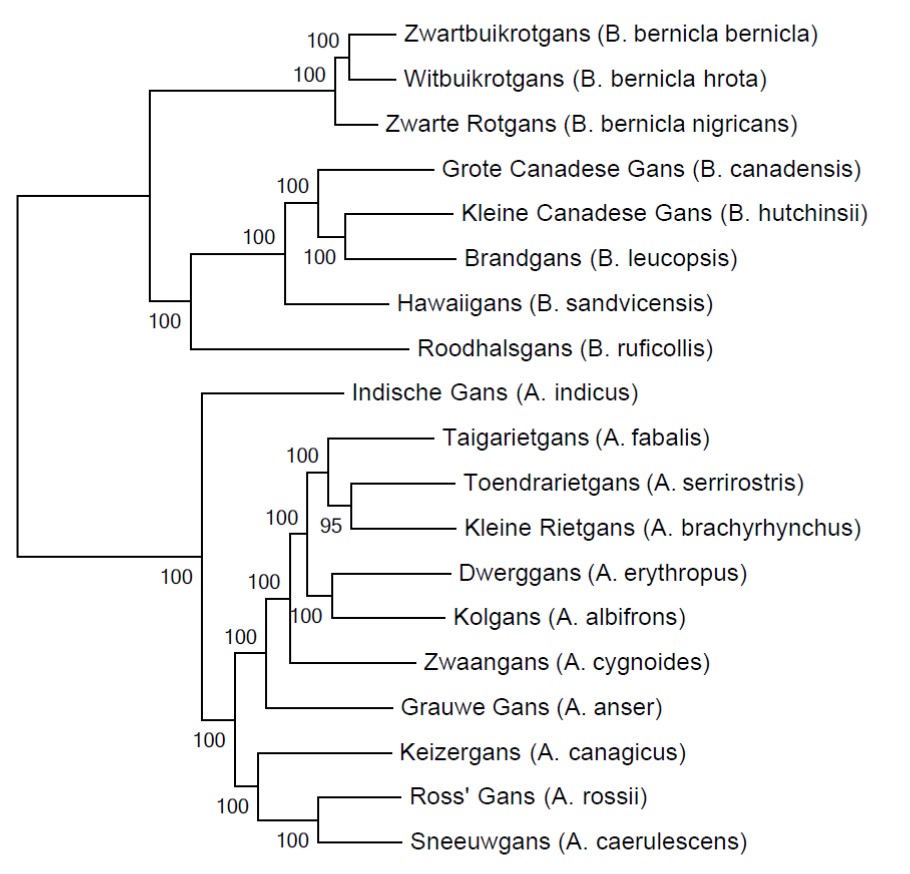
Betrouwbaarheid
Je kan relatief makkelijk de genetische afstanden berekenen en een evolutionaire boom tekenen. Maar hoe betrouwbaar zijn die resultaten? In de jaren 80 ontwikkelden wetenschappers (voornamelijk Joseph Felsenstein) statistische methoden om de betrouwbaarheid van evolutionaire bomen te schatten. De meeste gebruikte test is bootstrapping waarbij men willekeurig letters uit de DNA-sequenties kiest om vervolgens de analyses opnieuw te doen met deze willekeurige dataset. Dit proces wordt enkele duizenden keren herhaald. Uiteindelijk berekent men in hoeveel procent van de analyses dezelfde groepen in de evolutionaire boom gevonden werden. In de figuur hierboven zie je een voorbeeld van mijn ganzenonderzoek, gebaseerd op een DNA-sequentie van meer dan zes miljoen letters. De bootstrap-percentages zijn allemaal honderd procent, behalve de groep met Toendrarietgans en Kleine Rietgans (maar 95 procent is ook niet slecht).
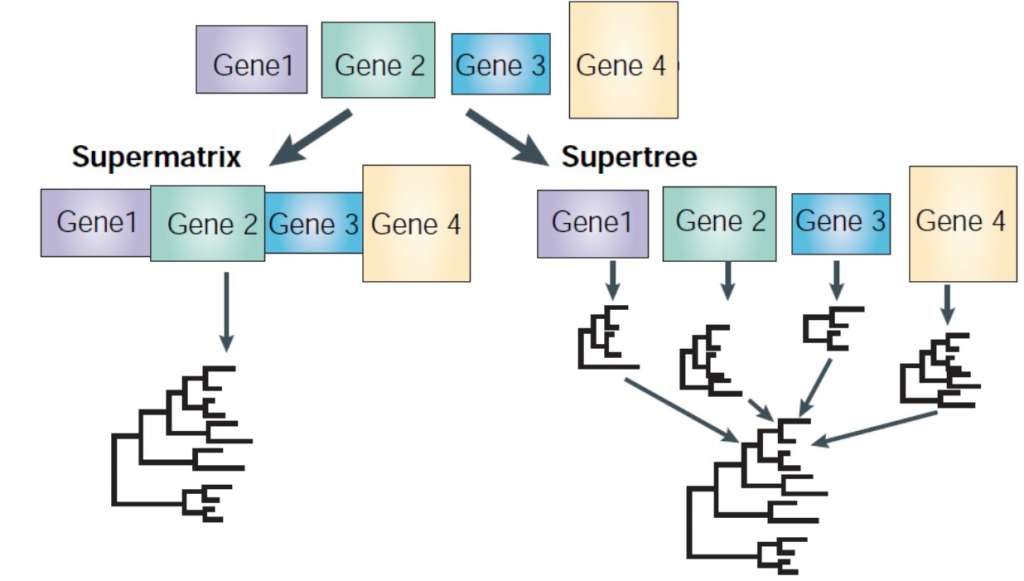
Sinds enkele jaren is het mogelijk om het volledig genoom van een individu in kaart te brengen. Wetenschappers wreven zich al in de handen, met zoveel data konden ze vast en zeker enkele lastige evolutionaire vraagstukken oplossen. Maar de analyses van genoomdata leidden tot een grote anticlimax. Verschillende genen resulteerden in verschillende evolutionaire bomen. Het werd dus alleen maar ingewikkelder. Momenteel passen wetenschappers twee strategieën toe. In de eerste strategie worden alle genen achter elkaar geplakt en geanalyseerd alsof het één groot gen is. De andere strategie omvat aparte analyses van alle genen waarna men alle gen-bomen tot een consensus stamboom combineert. Er bestaat nogal wat onenigheid onder evolutiebiologen over welke aanpak nu de beste is. Het debat is nog volop aan de gang.
Jente Ottenburghs promoveerde aan de Universiteit Wageningen waar hij onderzoek deed naar de evolutie van ganzen. Na een stage bij de wetenschapsredactie van de Volkskrant werkt hij nu als postdoc aan het Karolinska Institutet in Stockholm (Zweden). Meer weten over Jente? Neem een kijkje op zijn website. Recent kon je in een artikel van de hand van Jente al lezen hoe een genoom in kaart wordt gebracht. Nieuwsgierig? Klik hier! En hier kun je lezen hoe de genetische code precies werkt.



